Обновлено 03.05.2019
Болезнь Пелицеуса-Мерцбахера по своей сути является группой не до конца изученных демиелинизирующих заболеваний, при которой имеется грубый нистагм, атактические нарушения и аномалии в функционировании и строении миелиновой оболочки в белом веществе головного мозга. Заболевание передается наследственно, рецессивным путем X-сцепленное с полом, следовательно, болеют только мальчики, девочки являются носителями мутированных генов.
Код по МКБ 10 G37.8. Шифровка по МКБ 11 вероятнее всего будет осуществляться под кодом 8A44.0, в классе болезни нервной системы, рубрика демиелинизирующие заболевания, подрубрика лейкодистрофии, либо в разделе аномалий развития, под кодом LE12.
Содержание:
Причины
Болезнь Пелицеуса-Мерцбахера является наследственным заболеванием. Классически передается по рецессивному Х-сцепленному типу наследования (хромосома Xq22), вызывается аномалией гена, кодирующего синтез протеолипидного белка, структурно важного для формирования миелина в ЦНС, а также для дифференциации олигодендроцитов. Мутации на этом же гене могут приводить к развитию семейного спастического парапареза.
В настоящее время разработаны подходы к молекулярной диагностики заболевания с использованием мутационного анализа. Но несмотря на это, как и при ряде других заболеваний с Х-сцепленным типом наследования, диагностика болезни Пелицеуса-Мерцбахера имеет ряд затруднений, ведь мутации в экзонах выявляются лишь у 10-25 % пациентов с данным заболеванием.
Симптомы
Для болезни Пелицеуса-Мерцбахера характерна триада симптомов: грубый вертикальный и/или горизонтально-ротаторный нистагм, кивательные движения головы и нарушение координации. Заболевание дебютирует в младенчестве (5-10 месяцев) отличается медленным темпом прогрессированиея. Постепенно к вышеуказанной симптоматике добавляется повышение мышечного тонуса, атрофия зрительного нерва, брадилалия (замедление темпа речи), прогрессирующее снижение интеллекта. На поздних стадиях присоединяются симптомы Паркинсонизма, гииеркинезы, нарастает деменция. Болезнь прогрессирует особенно быстро в первые годы жизни. В дальнейшем иногда наблюдаются периодические ремиссии на протяжении длительного времени, однако без существенного регресса имеющихся симптомов.
Диагностика
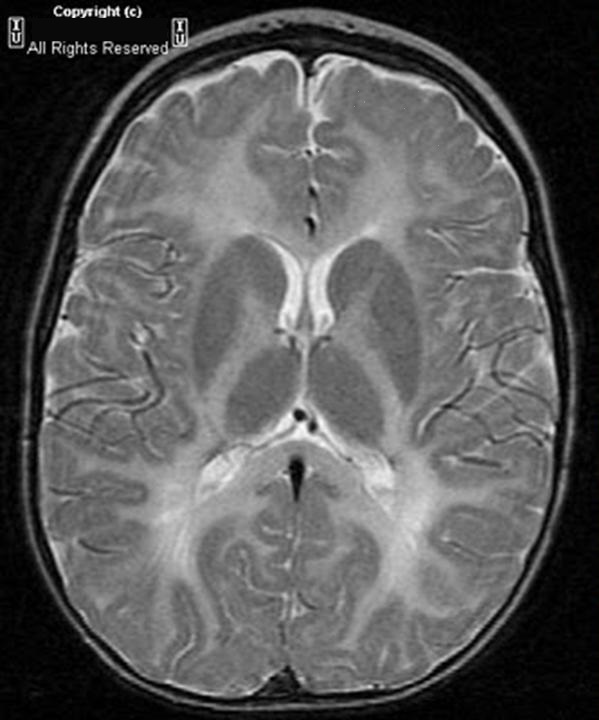
Диагноз основывается на выявлении характерной триады вышеупомянутых неврологических симптомов в младенчестве с их постепенным медленным нарастанием. В ликворе может выявляться незначительное повышение уровня белка, а также цитоз. Основные патоморфологические изменения со стороны нервной ткани включают отсутствие миелиновой оболочки неповрежденных аксонов, что предполагает под собой также нарушение функции олигодендроглии. Исследования показали существование генетического дефекта в биосинтезе протеолипида апопротеина, играющего роль в дифференцировки олигодендроцитов и поддержании их функционирования. На МРТ исследовании определяется симметричный паттерн замедления темпов миелинизации (см. фото). Исследование слуховых вызванных потенциалов ствола мозга на ранних этапах заболевания демонстрируют паттерн отсутствия волн III-V. При исследовании зрительных вызванных потенциалов отмечается замедление латентностей, соматосенсорные вызванные потенциалы показывают отсутствие кортикальных ответов, либо также замедление латентностей. Данные имеют большое значение при обследовании мальчиков при наличии у них нистагма, позволяя в большинстве случаев произвести полноценную дифференциальную диагностику заболевания.
Лечение
Патогенетического лечения болезни Пелицеуса-Мерцбахера не существует. Прибегают к общим методикам реабилитации. В ряде случаев возможно симптоматическое лечение.
Прогноз
Прогноз неблагоприятный. Заболевание всегда заканчивается летальным исходом чаще ко второму-третьему десятилетию жизни.
Источники литературы
Михайлова С. В., Захарова Е. Ю., Петрухин А. С. Нейрометаболические заболевания у детей и подростков. Диагностика и подходы к лечению. -М: Издательство «Литтерра», 2011.-С. 324-331.
Bonnefond L. et al. Towards the full set of human mitochondrial aminoacylt RNA synthetases: charakterization of AspRS and TyrRS//Biochemistry. -2005. -Vol. 44. -P. 4805-4816.
Paepe de B. et al. Diagnostic value of immuno-staining in cultured skin fibroblasts from patients with oxidative phosphorylation defects//Pediat. Res. -2006. -Vol. 59. -P. 2-6.
Huang Q. H., Xiao J. X., Wang J. M., Jiang Y. W, Wu Y. Clinical and genetic analysis of a family with leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation//Zhonghua Er Ke Za Zhi. -2012. -Vol. 50(1). -P. 50-55. Chinese.